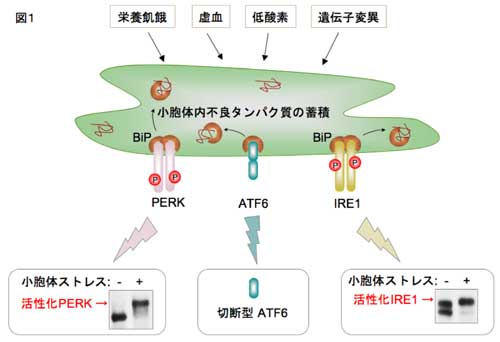
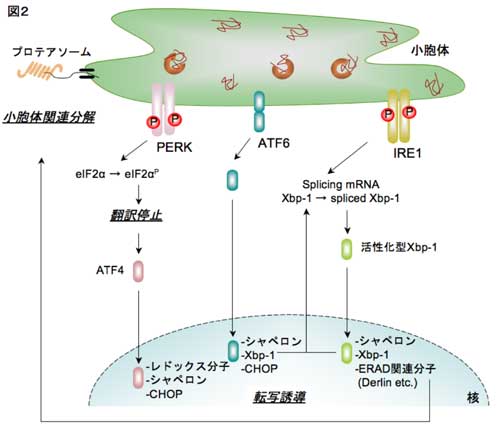
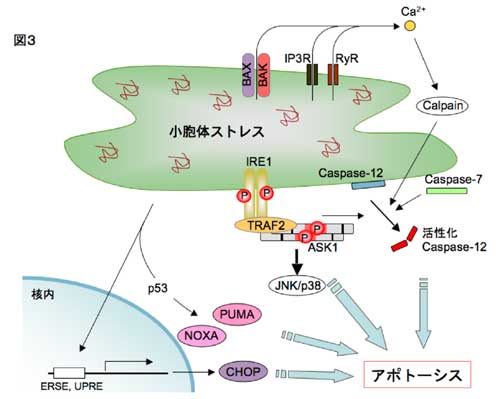
�R�D���E�̃X�g���X�Ɛ_�o�ϐ�����
�@�ߔN�A���E�̃X�g���X�Ɨl�X�Ȏ����i���A�a�E�����������E�_�o�ϐ������E�������Ȃǁj�̊W�����炩�ɂȂ��Ă��܂��B���̂Ȃ��Ŏ������́A�����_�o�ϐ������Ə��E�̃X�g���X�̊W�ɂ��ďœ_�����āA������i�߂Ă��܂��B
�E�|���O���^�~���a
�@�n���`���g���a��}�V���h���Z�t�a�Ȃǂ̃|���O���^�~���a�́A������`�q���ɂ�����CAG�̌J��Ԃ����̑����ɂ��|���O���^�~���^���p�N���������_�o�זE���ɒ~�ς��A�s�n���ÏW����`�����邱�Ƃɂ���Ĕ��ǂ����`���̐_�o�ϐ������ł��B�}�E�X����|�{�_�o�זE�Ƀ|���O���^�~���^���p�N���������邱�Ƃɂ��A���E�̃X�g���X����N����邱�ƁA�����ASK1-JNK�o�H����������������邱�Ƃ����炩�ɂȂ�܂����B�܂��AASK1-/-�_�o�זE��p���������ɂ��A�|���O���^�~���U�����_�o�זE��������ASK1�̕K�v����������܂����B����A���E�̂̊O�ɑ��݂���|���O���^�~���^���p�N�����A�ǂ̂悤�ɂ��ď��E�̃X�g���X��U�����邩�ɂ��ẮA���̒~�ςɂ���ăv���e�A�]�[�������̒ቺ���ώ@����Ă���A����ɂ��ERAD��������E�̓��s�ǃ^���p�N���̕������}������A���ʓI�ɏ��E�̓��o�Ɉُ�^���p�N�������X�ɒ~�ς���ƍl�����܂��iNishitoh et al. Genes Dev. 2002�j�B
�E�؈ޏk�������d����
�@
�؈ޏk�������d���ǁiAmyotrophic lateral sclerosis�GALS�j�́A�^���_�o�����ٓI�ɏ�Q����邫��߂ďd�ĂȐi�s���_�o�ϐ������ł��B���{�����̜늳�Ґ��͖�7000�l�B���݂̎��Ö@�́A���n�r���e�[�V�����ɂ��ؗ͒ቺ�\�h�ƁA�O���^�~���_�_�o�I�����o�}���܂ɂ��ꎞ�I�Ȑi�s�x�������ŁAALS�a�ԕ��q���J�j�Y���Ɋ�Â����{�I�Ȏ��Ö@�͂���܂���B�Ƒ���ALS�̌����Ƃ���1993�N�ɔ������ꂽCu/Zn superoxide dismutase 1�iSOD1�j�̈�`�q�ψق́A�Ƒ��������łȂ��ǔ���ALS�̕a�ԉ𖾂ɂ�����I�Ȑi���������炷�Ɗ��҂���Ă��܂����A����܂Ŏ��ÕW�I�ƂȂ镪�q���J�j�Y���͖��炩�ɂ���Ă��܂���ł����B�������́AALS�̕a�ԕ��q���J�j�Y���Ƃ��āAERAD�̋@�\�ቺ�ɂ�鏬�E�̃X�g���X�U�����A�|�g�[�V�X�̊֗^�𖾂炩�ɂ��܂����iNishitoh et al. Genes Dev. 2008�j�B
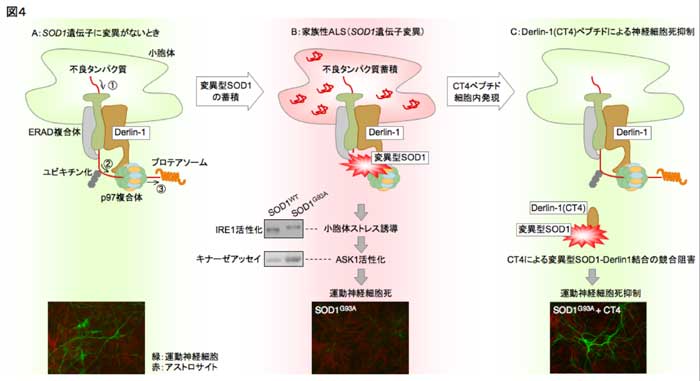
�}�S�F�ψٌ^SOD1�ɂ�鏬�E�̃X�g���X�U���Ɖ^���_�o�זE���F�@(A)���E�̊֘A�����iERAD�j�̗���(B)�ψٌ^SOD1��Derlin-1�Ɍ������AERAD��̗����j�Q���A���̌��ʏ��E�̃X�g���X�U������ASK1����������N����A�^���_�o�זE�����N����B�����̃p�l����IRE1�������ɂ�郊���_����ASK1�̊������iin vitro�L�i�[�[�A�b�Z�C�j���A���̃p�l���͕ψٌ^SOD1�ɂ��^���_�o�זE���������B(C)�ψٌ^SOD1�����̈��12�A�~�m�_CT4�́A�ψٌ^SOD1-Derlin-1�����������j�Q���^���_�o�זE����}������B
�@i) �ψٌ^SOD1�ɂ�鏬�E�̊֘A�����iERAD�j�̗}��
�@���E�̓��s�ǃ^���p�N���́A���E�̖����t�A������i�}�SA,�@�j�A������E3���r�L�`�����K�[�[�ɂ���ă|�����r�L�`�����i�A�j�A�v���e�A�\�[���ɂ���ĕ�������܂��i�B�j�B����ERAD��^���p�N���̋t�A���ߒ��ɂ́A���E�̖��ɑ��݂���Derlin�t�@�~���[���q���֗^���Ă��܂��B�^���_�o�n�iNSC34�j�זE�ɁA3��ނ̕ψٌ^SOD1�iSOD1A4V, SOD1G85R, SOD1G93A�j���������Ƃ���A���E�̎�e��IRE1�i�}�SB�j, PERK�̊��������ώ@����܂����BSOD1�͎�ɍזE�����ɔ������邽�߁A�ǂ̂悤�ɂ��ď��E�̓��o�ɕs�ǃ^���p�N����~�ς����邩���^��ł��������߁AERAD��^���p�N���̕��x�����������Ƃ���A�쐶�^SOD1�ɔ�ׂĕψٌ^SOD1�̋������ɂ���ėL�ӂɒx���������Ƃ���A�ψٌ^SOD1�͉��炩�̃��J�j�Y������āAERAD�o�H����ٓI�ɗ}�����Ă��邱�Ƃ���������܂����B
�@ii) �ψٌ^SOD1��Derlin-1�̓��ٓI����
�@�ψٌ^SOD1�́A�l�X�ȕ��q�ƌ������邱�Ƃ�����Ă��܂��B�����ŁAERAD�}���ɂ�ERAD�����́i�}�SA�j�ƕψٌ^SOD1�̌������֗^���Ă���Ɨ\�z���A�����̌�����in vitro�Ō��������Ƃ���ADerlin-1�Ƃ���4�ђʌ^���q�Ɍ������A���̌�����NSC34�זE�Ȃǂ̔|�{�זE�ASOD1G93A�g�����X�W�F�j�b�N�iALS�j�}�E�X�Ґ���p���������ł��m�F����܂����B�܂��A����Derlin�t�@�~���[���qDerlin-2, -3�ɂ͑S���������Ȃ��������ƂȂǂ���A�ψٌ^SOD1��ERAD�����̕��q�̂Ȃ��ŁA����߂ē��ٓI��Derlin-1�Ɍ�������Ƃ����܂��B���̌����̈�Ɋւ���ڍׂȌ����̌��ʁADerlin-1(C���[)��12�A�~�m�_�̈�肵�܂����B
�@iii) Derlin-1�̊l�����@�\��Q
�@Derlin�t�@�~���[�͓��肳�ꂽ�����A�t�A���E�i���g���g�����X���R���j���\�����镪�q�̈�Ɨ\�z����܂������A���݂܂Ŗ��m�Ȏ����I�؋��͓����Ă��炸�AERAD�̋@�\�ɕK�v�ȕ��q�ł��邱�Ƃ͎�����Ă�����̂́A�ǂ̂悤�Ȗ�����S���Ă��邩�͕s���ł��B�ψٌ^SOD1�́AERAD��̃��r�L�`�����������}���������Ƃ���A�ψٌ^SOD1��ERAD�́u����v��}�����A���̌��ʏ��E�̕s�ǃ^���p�N����~�ς����Ă���Ɨ\�z����܂����i�}�SB�j�B�ψٌ^SOD1��Derlin-1�̋@�\��P�ɑr�������Ă���̂ł͂Ȃ��A�l�����@�\�ُ�������N�������ƂŁA���E�̃X�g���X��U�����Ă��܂��B���݂́A�ψٌ^SOD1�����������ǂ̂悤��Derlin-1�@�\��Q�������N�����Ă���̂��𖾂炩�ɂ��A����ɂ���ɂ����Derlin-1�̐����I�@�\���𖾂��悤�Ǝ��݂Ă��܂��B
�@iv) ���E�̃X�g���X-ASK1�o�H������^���_�o�זE��
�@
�ψٌ^SOD1�ˑ��I�ɏ��E�̃X�g���X���U������邱�Ƃ���AASK1�o�H�ɂ��Č��������Ƃ���A TRAF2-ASK1�̌����ƁA����ɔ���ASK1�̊��������ώ@����܂����i�}�SB�j�B���̌o�H���A�^���_�o�זE���Ɋ֗^���邩�ۂ����������邽�߁A�}�E�X�َ��̐Ґ��|�{�n��p���Ă̕ψٌ^SOD1�ˑ��I�^���_�o�זE�������n�Ɂi�}�SB�j�A�ψٌ^SOD1-Derlin-1������j�Q����CT4�y�v�`�h�i12�A�~�m�_�j���������Ƃ���A�^���_�o�זE�����L�ӂɗ}�������ƂƂ��Ɂi�}�SC�j�AASK1�m�b�N�A�E�g�}�E�X�R���̍זE�ł����l�̌��ʂ������܂����B����ɁAALS�}�E�X��ASK1-/-�}�E�X�̌�z�������s�������ʁAALS�}�E�X�ł݂���Ґ��O�p���^���_�o�זE�ϐ���ASK1-/-�}�E�X�ŗ}������A�������Ԃ̗L�ӂȉ������݂��܂����B
�@�]���āA�ψٌ^SOD1��Derlin-1�̌�����������E�̃X�g���X�U����ASK1���������^���_�o�זE����U�����A�����̃��J�j�Y����ALS�̕a�Ԑi�s�ɐ[���ւ���Ă��邱�Ƃ�������܂����B
|